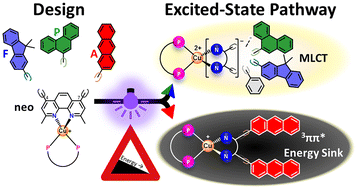
A modular design approach for bichromophoric Cu(I) complexes is reported, combining a [Cu(N^N)(P^P)]+ scaffold that supports metal-to-ligand charge transfer with two polycyclic aromatic hydrocarbon chromophores covalently attached to the diimine ligand. Three new systems incorporating 9,9-dimethyl-9H-fluorene, phenanthrene, and anthracene units at the 4,7-positions of neocuproine were synthesized and fully characterized. Structural analysis reveals twisted geometries of the attached chromophores, which reduce conjugation with the diimine core. Electrochemical measurements indicate only moderate shifts in reduction potentials, indicating preservation of the Cu(I)-centered redox behavior. Across the series, the visible MLCT band is retained without pronounced red-shifts, while the molar attenuation coefficients increase markedly (×1.7–3.4 vs. the reference), in line with calculated oscillator strengths. Fragment comparisons and computations attribute the additional UV intensity to intraligand charge transfer (9,9-dimethyl-9H-fluorene, phenanthrene) and to vibronically structured πA* ← πA transitions (anthracene), consistent with the non-coplanar geometries. Complexes bearing 9,9-dimethyl-9H-fluorene and phenanthrene substituents exhibit yellow emission involving the Cu(I)-based metal-to-ligand charge transfer state (λem ≈ 565 nm, τ up to 20.8 μs), whereas the anthracene-substituted system populates a non-emissive 3π–π* triplet state localized on an anthracene moiety (τ > 35 μs), as evidenced by transient absorption spectroscopy and time-dependent density functional theory. These results establish clear structure–property relationships in bichromophoric Cu(I) systems and illustrate how polycyclic aromatic hydrocarbon chromophores influence excited-state character and dynamics.