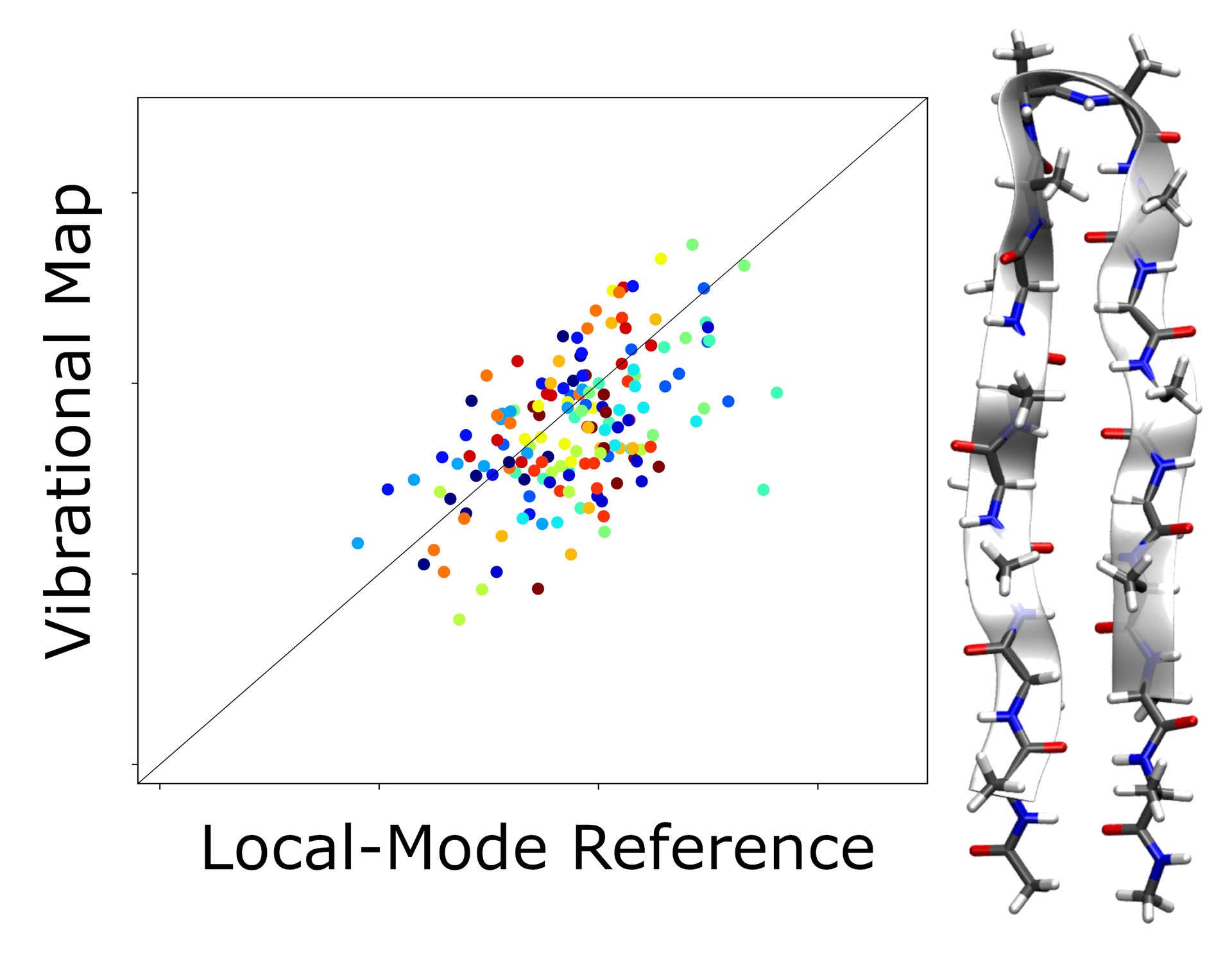
Vibrational exciton models are widely used for the simulation of biomolecular vibrational spectra, in particular of two-dimensional infrared (2D-IR) spectra. The parameters entering such models, specifically local-mode frequencies and coupling constants, are provided by vibrational maps, which have been parametrized agains computational data for small molecules as well as experimental data. Here, we put forward a novel approach for assessing the quality of these vibrational maps against quantum-chemical reference data. For a test set consisting of molecular dynamics snapshots of polypeptides and small proteins, covering different secondary structure motifs, we we performed full quantum-chemical calculations of harmonic vibrational frequencies and normal modes, and applied a localization of normal modes to obtain localized-mode frequencies and coupling constants. These can be directly compared to those predicted by vibrational maps. We find that while there is a good correlation for the coupling constants and for local-mode frequencies of isolated polypeptides, there is hardly any correlation for the local-mode frequencies of solvated polypeptides. This striking finding calls into question the accuracy of the electrostatic maps that are used to model the effect of the solvent molecules on local-mode frequencies.