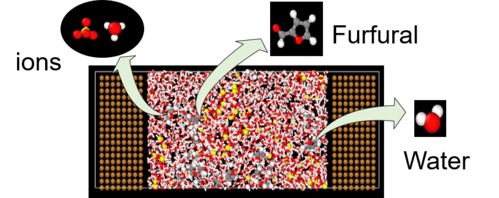
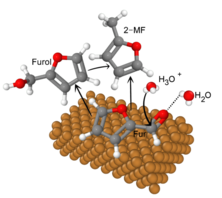
Im SE2A Cluster werden detaillierte MD Simulation zum Reaktionssystem der Elektrosynthese alternativer Kraftstoffe durchgeführt , um ein vertieftes Verständnis der Elektrolyt-Elektroden Wechselwirkungen gewinnen zu können. Dabei sollen Ad- und Desorptionsverhalten der Edukte und Produkte an der Elektrodenoberflächen, ihre mikrostrukturellen Eigenschaften sowie ihr Diffusionsverhalten in Lösung in Abhängigkeit vom Elektrodenmaterial, der Elektrolytzusammensetzung, der Stromstärke u.ä. analysiert werden. Die Simulationsergebnisse werden in Relation zu experimentellen Beobachtungen gesetzt, um Korrelationen zwischen strukturellen und dyamischen Eigenschaften der Komponenten und bevorzugten Reaktionspfaden zu ermitteln. Durch die Identifizierung der relevanten molekularen Einflussfaktoren sollen Strategien für maßgeschneiderte Elektrosyntheseprozesse abgeleitet werden, die eine hohe Reaktionsrate, Ausbeute und Energieeffizienz ermöglichen.
M. Sc. Sahar Rabet