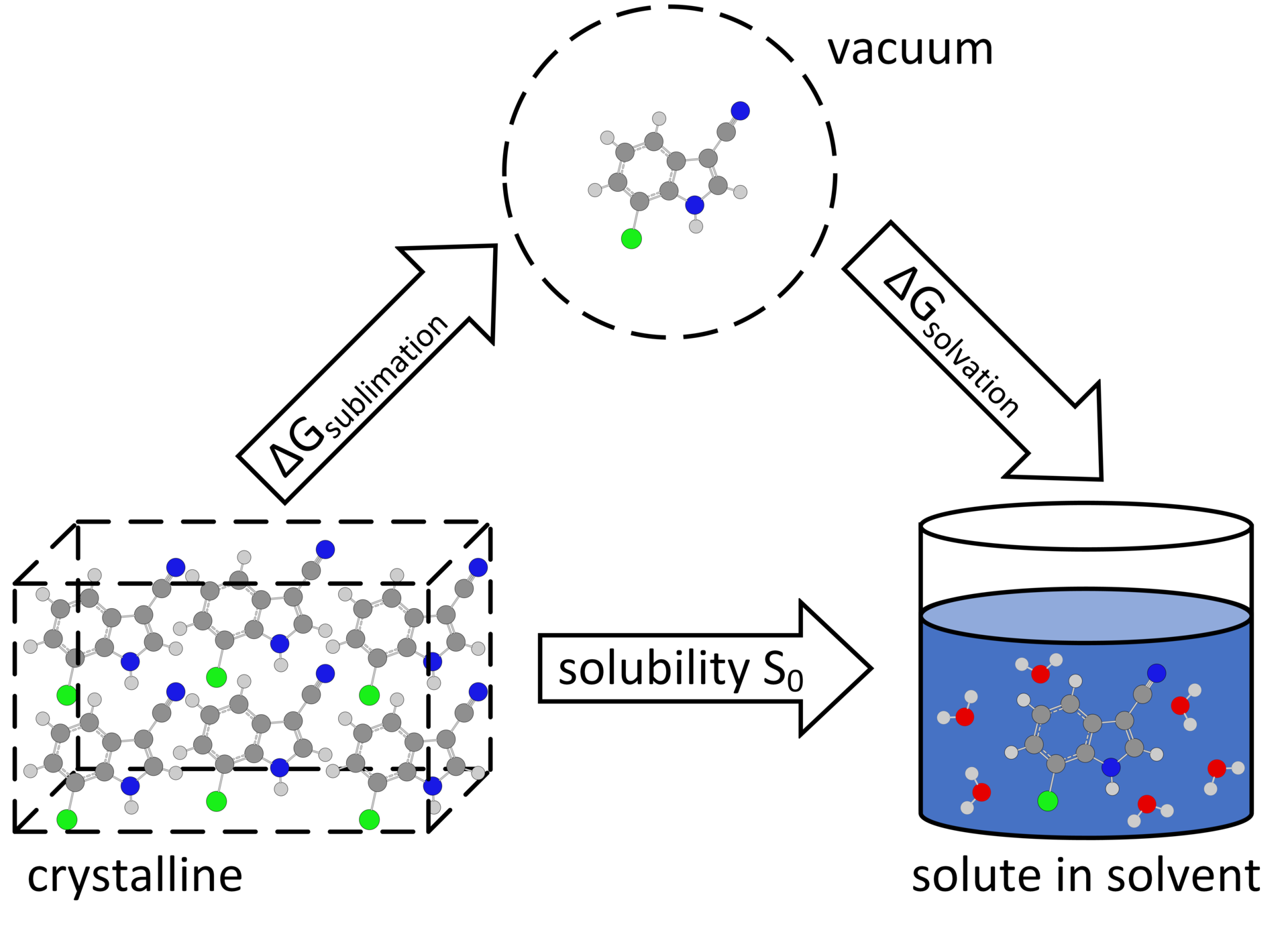
Solubility is an import thermophysical property of potential active pharmaceutical ingredients (API) as it affects the intake and availability in the organism. Molecular simulations allow for the calculation of relative solubilities and enable detailed insights into the solubility processes on the molecular level. This can help to specifically design drugs with improved solubility or to recommend suitable solvents for the synthesis or analytics of medicaments. The working group Molecular Thermodynamics supports the research activities of the Center of Pharmaceutical Engineering (PVZ) at TU Braunschweig, aiming at the cost efficient development of personalized drugs. The focus of our work is the reduction of the high computational effort necessary for the solubility calculations and the optimization of molecular models.
Subject in our new project within the Lower Saxony doctoral program "Drug Discovery und Chemieinformatik für neue Antiinfektiva (iCA) is the advancement of simulation methods and force field models for solubility studies of organometallic compounds. The parameterization of the force field models shall be based on ab initio simulations, which yield additional information, such as bonding geometries, charge distribution etc. Additionally, molecular simulation studies will be performed to analyze the solvation structures of organometallic compounds. The information from both, ab initio and molecular simulations, will be related to the biological activity of the organometallic compounds to allow for a computer-aided rational drug design.